
Expo
view channel
view channel
view channel
view channel
view channel
view channel
Medical Imaging
AI
Surgical TechniquesPatient CareHealth ITPoint of CareBusiness
Events
Webinars

- FDA-Cleared Index Enables Pediatric-Specific Sedation Monitoring
- Smart Ring Continuously Monitors Glucose and Ketones from Passive Sweat
- Invisible Skin Sensors Could Transform Everyday Biosignal Monitoring
- Patient-Specific Artery-on-a-Chip Refines Stroke Risk Stratification
- Existing Cardiovascular Risk Calculators Predict Several Common Cancers
- AI Analyzes Surgical Movements to Improve Prostatectomy Outcomes
- Photostable NIR Dye Sustains Intraoperative Imaging in Cancer Surgery
- Next-Generation Total Artificial Heart System to Be Presented at IEEE EMBC 2026
- Single Robotic Platform Supports Three Minimally Invasive Approaches
- Navigation-Compatible Instruments Improve Precision in Lumbar Facet Fusion
- Wearable Sleep Data Predict Adherence to Pulmonary Rehabilitation
- Revolutionary Automatic IV-Line Flushing Device to Enhance Infusion Care
- VR Training Tool Combats Contamination of Portable Medical Equipment
- Portable Biosensor Platform to Reduce Hospital-Acquired Infections
- First-Of-Its-Kind Portable Germicidal Light Technology Disinfects High-Touch Clinical Surfaces in Seconds
- Danaher Completes Acquisition of Masimo to Expand Patient Monitoring Capabilities
- Endologix Adds FDA-Cleared Peripheral Thrombectomy System
- Artivion Adds FDA-Approved NEXUS System to Aortic Arch Portfolio
- Olympus Partnership Aims to Expand Access to Robot-Assisted Endoscopic Therapy
- Johnson & Johnson Launches AI-Driven Cardiac Mapping System
- Smartwatch Monitoring Identifies Abnormal Vital Signs Before Routine Checks
- Weekly Remote Symptom Monitoring Improves Symptom Control in Advanced Cancer
- Digital Heart Model Supports Targeted Ablation in Atrial Fibrillation
- AI Framework Helps Clinicians Create Trustworthy Risk Prediction Tools
- AI Tool Screens for Primary Aldosteronism Using Routine EHR Data
- Handheld Ultrasound Expands Point-of-Care Imaging Access in Brazil
- AI Dermatology Platform Targets Early Detection of Non-Melanoma Skin Cancer
- Handheld AI Device for Point-of-Care Skin Lesion Assessment Receives CE Mark
- Portable Immunoassay System Advances Toward Point-of-Care Biomarker Testing
- Portable MRI System Accelerates Emergency Brain Imaging and Triage
- FDA-Cleared Index Enables Pediatric-Specific Sedation Monitoring
- Smart Ring Continuously Monitors Glucose and Ketones from Passive Sweat
- Invisible Skin Sensors Could Transform Everyday Biosignal Monitoring
- Patient-Specific Artery-on-a-Chip Refines Stroke Risk Stratification
- Existing Cardiovascular Risk Calculators Predict Several Common Cancers
- AI Analyzes Surgical Movements to Improve Prostatectomy Outcomes
- Photostable NIR Dye Sustains Intraoperative Imaging in Cancer Surgery
- Next-Generation Total Artificial Heart System to Be Presented at IEEE EMBC 2026
- Single Robotic Platform Supports Three Minimally Invasive Approaches
- Navigation-Compatible Instruments Improve Precision in Lumbar Facet Fusion
- Wearable Sleep Data Predict Adherence to Pulmonary Rehabilitation
- Revolutionary Automatic IV-Line Flushing Device to Enhance Infusion Care
- VR Training Tool Combats Contamination of Portable Medical Equipment
- Portable Biosensor Platform to Reduce Hospital-Acquired Infections
- First-Of-Its-Kind Portable Germicidal Light Technology Disinfects High-Touch Clinical Surfaces in Seconds
- Danaher Completes Acquisition of Masimo to Expand Patient Monitoring Capabilities
- Endologix Adds FDA-Cleared Peripheral Thrombectomy System
- Artivion Adds FDA-Approved NEXUS System to Aortic Arch Portfolio
- Olympus Partnership Aims to Expand Access to Robot-Assisted Endoscopic Therapy
- Johnson & Johnson Launches AI-Driven Cardiac Mapping System
- Smartwatch Monitoring Identifies Abnormal Vital Signs Before Routine Checks
- Weekly Remote Symptom Monitoring Improves Symptom Control in Advanced Cancer
- Digital Heart Model Supports Targeted Ablation in Atrial Fibrillation
- AI Framework Helps Clinicians Create Trustworthy Risk Prediction Tools
- AI Tool Screens for Primary Aldosteronism Using Routine EHR Data
- Handheld Ultrasound Expands Point-of-Care Imaging Access in Brazil
- AI Dermatology Platform Targets Early Detection of Non-Melanoma Skin Cancer
- Handheld AI Device for Point-of-Care Skin Lesion Assessment Receives CE Mark
- Portable Immunoassay System Advances Toward Point-of-Care Biomarker Testing
- Portable MRI System Accelerates Emergency Brain Imaging and Triage




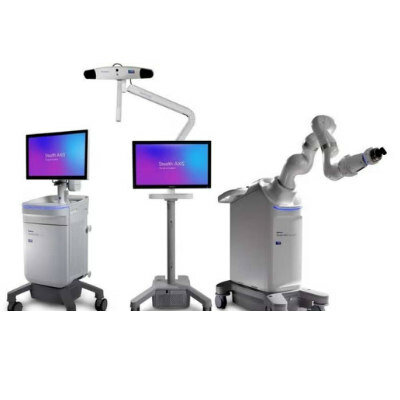

 technologies, defining a new category in medicine and revolutionizing neurorecovery. (Photo courtesy of Neuvotion)")
")
")
")
")
")
 and its enhanced photostability. (Photo courtesy of Korea Research Institute of Chemical Technology)")
 ventricles with the next-generation electromechanical Emperor Drive System (Photo courtesy of Picard Medical)")
")
")
")
")
")


. DOI: 10.1113/jp290765)")
. (Photo courtesy of Longeviti Neuro Solutions)")
")
")
")
")



